РОССИЙСКАЯ АКАДЕМИЯ НАУК ИНСТИТУТ ЦИТОЛОГИИ
А. А.МИРОНОВ, Я. Ю. КОМИССАРЧИК, В. А.МИРОНОВ
МЕТОДЫ ЭЛЕКТРОННОЙ МИКРОСКОПИИ В БИОЛОГИИ И МЕДИЦИНЕ
Ответственный редактор Н. Н. Никольский
ПРЕДИСЛОВИЕ
1.1. ВЗЯТИЕ МАТЕРИАЛА
1.2. ХИМИЧЕСКАЯ ФИКСАЦИЯ
1.3. ХИМИЧЕСКИЕ ФИКСАТОРЫ
1.3.1. Характеристика фиксаторов
1.3.1.1. Четырехокись осмия (ЧО)
1.3.1.1.1. Режим использования
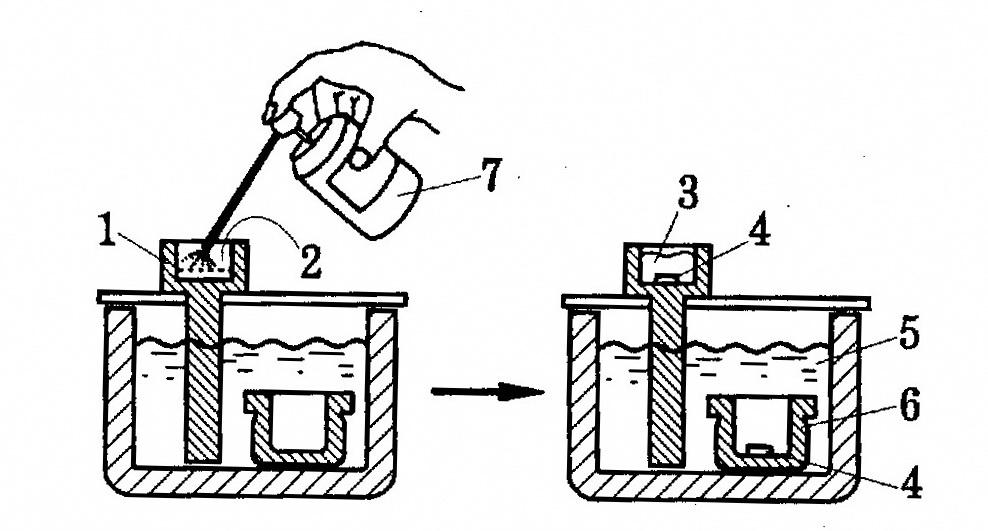
1.3.1.1.2. Приготовление растворов
1.3.1.2. Глютаральдегид (ГА)
1.3.1.2.1. Механизмы действия
1.3.1.2.2. Режим использования
1.3.1.2.3. Состав коммерческих растворов ГА
1.3.1.3. Формальдегид (ФА)
1.3.1.4. Акролеин
1.3.2. Выбор фиксаторов
1.3.3. Буферные растворы
1.3.4. Осмотическое давление
1.3.4.1. Промывание
1.3.5. Способы химической фиксации
1.3.5.1. Перфузионная фиксация
1.3.5.1.1. Отмывание
1.3.5.1.2. Введение фиксатора
1.3.5.2. Иммерсионная фиксация
1.3.5.3. Инъекция
1.4. МИКРОВОЛНОВОЕ ИЗЛУЧЕНИЕ (МВИ)
1.4.1. Механизмы действия МВИ
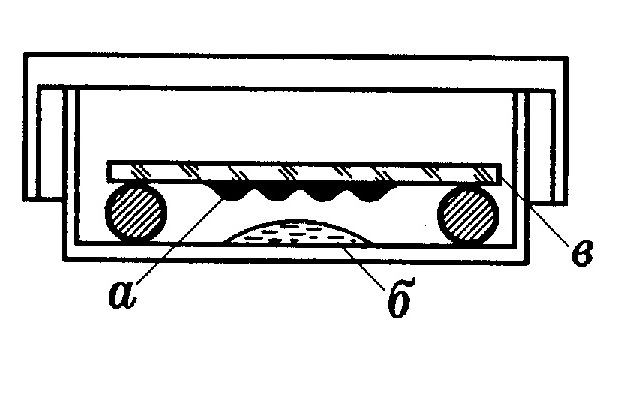
1.4.2. Устройства для МВИ
1.4.3. Режимы фиксации
1.4.4. Температурный режим фиксации
1.5. ФИКСАЦИЯ И ИММУНО-ЭМ
1.5.1. Требования к фиксации
1.5.2. Блокирование остаточных альдегидных групп
1.6. ОСОБЕННОСТИ ФИКСАЦИИ КУЛЬТИВИРУЕМЫХ КЛЕТОК
1.7. ХРАНЕНИЕ ФИКСИРОВАННОГО МАТЕРИАЛА
1.8. АРТЕФАКТЫ 1.8.1. Артефакты, возникающие при взятии материала
1.8.2. Артефакты, возникающие при фиксации
1.8.3. Артефакты, связанные с иммерсионной фиксацией
1.8.4. Артефакты, обусловленные постфиксацией
2.1. МЕХАНИЗМЫ ЗАМОРАЖИВАНИЯ БИОЛОГИЧЕСКИХ ОБЪЕКТОВ
2.2. МЕТОДЫ БЫСТРОГО ЗАМОРАЖИВАНИЯ
2.2.1. Использование криогенов